Study opens new chapter on practice of single-cell genome de novo assembly
Jul 28, 2022
Peking University, July 28, 2022: With the increase in single-base accuracy and read length, single-molecule long-read sequencing technologies using bulk samples have been widely used in genome assembly.
Usually, the long-read sequencing assembly requires large amounts of DNA (typically several micrograms from millions of cells), and therefore most human genome assembly has been restricted to bulk genome sequencing datasets without keeping the potential genetic heterogeneities among individual cells. However, this is impractical for many situations.
By comparison, single-cell whole-genome sequencing (scWGS) is a powerful tool to reveal cell to cell genetic heterogeneities, especially for cancer research. Genomic assembly, or haplotype assembly, can be more accurate by using clonal populations that are more genetically similar.
Single-cell NGS genome sequencing technologies are commonly used in microbial genome assembly. However, NGS platform-based single cell genome sequencing technologies are rarely used in large and complex genome assembly, and even using bulk NGS genome sequencing data the assembly continuity of the contig N50 cannot achieve the megabase level, such as de novo assembly of human genome using bulk Illumina sequencing data alone with SOAPdenovo, yielding a contig N50 of 11.1 kb; de novo genome assembly for the American pika with contig N50 of ∼42 kb (using the Illumina HiSeq X (PE150bp) platform); de novo genome assembly for the American bison with contig N50 of ∼20 kb (using the Illumina and 454 paired-end libraries).
Assembling the human genome using a small amount of genomic DNAs or even a single cell genome sequencing data is much more challenging. It requires not only the support of single-cell TGS platform-based genome sequencing technology, but also a good data analysis strategy and a suitable assembler.
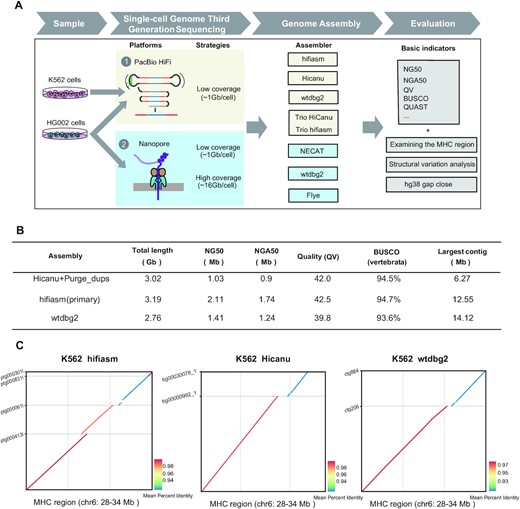
On July 12, a research team led by Prof. Tang Fuchou, at the School of Life Sciences, Biomedical Pioneering Innovation Center, Peking University, published an article in Nucleic Acids Research entitled De novo assembly of human genome at single-cell levels, in which they reviewed the team’s development of SMOOTH-seq technology, which can sequence the genome of a single-cell on the third-generation sequencing platform.
Figure 1
SMOOTH-seq can reliably and effectively detects SVs and ecDNAs in individual human cells, which made it possible to sequence a single cell genome with long reads (around 6kb), providing the prerequisite for human genome assembly from just several individual cells.
The team employed SMOOTH-seq on PacBio HiFi and Oxford Nanopore Technologies (ONT) platforms to sequence K562 (a human chronic myelogenous leukemia (CML) cell line) and HG002 (a normal diploid lymphoblast cell line) and demonstrate the feasibility of genome assembly based on scWGS dataset with different assemblers and rigorous evaluations (Figure 1A).
The team led by Prof. Tang Fuchou have systematically explored the factors that affect the assembly with TGS platform-based single-cell genome sequencing data. Furthermore, to investigate the lower limit of numbers of single cells need to be sequenced for genome assembly, the team improved the SMOOTH-seq technology (see methods), and sequenced 30 diploid HG002 cells with relatively high genome coverage on ONT platform, and found that the genome assembly from as low as 30 individual cells (average genome coverage ∼41.7%) can achieve NG50 of ∼1.35 Mb.
In addition, through analyzing the structural variations (SVs) of the assembled genome of K562 cells, the team found that compared with directly mapping single cell genome sequencing data onto reference genome, many more insertion events could be identified and complex structural variations could be more efficiently and accurately illustrated. Their research gave proof-of-principle evidences to show that it is feasible to assembly human genome with megabase level of NG50 contigs from long-reads single cell genome sequencing data of just a few dozens of individual cells.
The research paper was co-authored by Xie Haoling, a doctoral candidate at the School of Life Sciences, Peking University, and Li Wen, a doctoral candidate at the Academy for Advanced Interdisciplinary Studies of Peking University, with Prof. Tang Fuzhou as its corresponding author, and was supported by Center for Life Sciences (CLS), National Institute of Biological Sciences, Beijing Municipal Science and Technology Commission, and Beijing Advanced Innovation Center of Genomics (ICG).
Source: Biomedical Pioneering Innovation Center